
Sélectionnez votre langue
")
")
Le médecin investigateur peut vous proposer de participer à un essai clinique. Il vous délivre une information qui doit être claire, loyale et objective. Elle est d'abord orale et complétée par une note d'information écrite accompagnée du formulaire de consentement éclairé.
Ce formulaire de consentement « éclairé », signé par le patient, est obligatoire pour pouvoir participer à un essai clinique.
En principe, vous disposerez d'un délai de réflexion (1 semaine en général) compatible avec la sécurité et la qualité des soins, pour prendre votre décision et faire votre choix.
Vous devez être clairement informé :
Attention à ce stade, votre participation à l'essai clinique n'est pas encore certaine. En effet, chaque patient acceptant de participer à un essai clinique doit remplir un certain nombre de conditions (critères d'inclusions et de non inclusions). Ces critères sont déterminés par le protocole de l'essai. Ils permettent de recruter une population relativement homogène de participants et d'assurer le maximum de sécurité de ces derniers par rapport aux traitements proposés dans l'essai.
Tout au long de l'essai, le médecin se doit de délivrer cette information de qualité et s'efforcer de répondre à vos questions.
Vous avez le droit de :
Pour en savoir plus :
La participation à un essai clinique comprend le plus souvent : une phase de sélection puis une phase dite « de traitement » et enfin une phase de surveillance ou de suivi.
Le bénéfice attendu lié à votre participation à l'essai ainsi que le calendrier de l'essai qui permet d'évaluer le poids des contraintes ( durée et fréquence des visites, examens, tâches à effectuer au domicile, hospitalisation, etc.) doivent vous aider à prendre votre décision.
Cette phase commence avec la signature du formulaire de consentement. Elle vise à vérifier qu'il n'existe aucune contre-indication à la participation à l'essai (vérification des critères d'inclusion et de non-inclusion). Attention : la participation à la recherche commence avec la signature du formulaire de consentement, et non avec l'administration des traitements. Aucun examen de la recherche ne peut être effectué avant la signature du formulaire de recueil du consentement.
Si aucune contre-indication n'a été constatée, la phase dite de « traitement » commence. Il s'agit de la période pendant laquelle les participants vont recevoir les produits (produits à l'étude, traitements de référence, vaccins, etc.).
Le patient bénéficie d'une surveillance rapprochée tout au long de cette phase de traitement.
Après la dernière administration des produits, la phase de surveillance et de suivi permet de contrôler l'état de santé du participant. Cette phase varie selon les essais, d'une seule visite de contrôle, à plusieurs mois voire des années de surveillance, notamment en cas d'effets indésirables ou d‘évaluation à long terme comme pour les traitements adjuvants dans le cancer du sein.
Il est important de comprendre que l'arrêt du traitement ne signifie pas obligatoirement sortie de l'essai.
L'administration du traitement à l'étude peut être arrêté comme prévu au protocole de l'étude mais le patient reste dans l'étude pour la période de suivi pouvant inclure des consultations et des examens, décrits dans la note d'information. En général, le patient est suivi au minimum 30 jours après la dernière administration du produit à l'essai pour une évaluation de la tolérance.
On peut distinguer 2 types principaux de raisons de sortie d'essais :
Source La Ligue contre le cancer
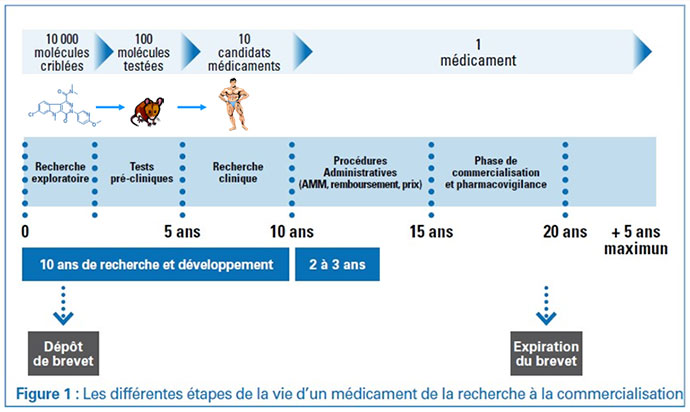
Tout essai clinique doit reposer sur des arguments scientifiques et éthiques valables, et surtout avoir pour but d'encadrer la participation des patients avec le minimum de risques.
En France, la recherche clinique est encadrée depuis 1988 par la Loi Huriet-Sérusclat. Elle organise, sécurise les essais, protège et informe les patients en spécifiant le rôle des différents acteurs et les droits des patients dans les essais.
La Loi Huriet-Sérusclat a été revue par la suite en 1990, en 1995 et dernièrement en août 2004.
Applicable à partir de la mi-2016, le nouveau règlement européen sur les essais cliniques de médicaments vise à mieux coordonner et faciliter les autorisations préalables des essais. Il assure également une plus grande transparence sur les résultats de ceux-ci.
Tout essai nécessite la collaboration de plusieurs acteurs :
Le promoteur propose le nouveau traitement, à partir d'un rationnel scientifique documenté (données expérimentales, besoin médical..). Le promoteur est la personne physique, la société ou l'institution qui propose de mettre en œuvre un essai thérapeutique et qui assure son financement. Il est responsable devant les autorités administratives du bon déroulement de l'essai thérapeutique et de la diffusion de toute information relative à la sécurité des personnes se prêtant à la recherche.
Les promoteurs peuvent être un groupe académique, une association de recherche médicale, l'industrie pharmaceutique, un médecin..,
Le promoteur rédige un protocole détaillé de l'essai ainsi que la note d'information et le consentement éclairé qui doivent être remis au patient. Ces documents sont ensuite soumis à un Comité de Protection des Personnes (CPP) et à l'Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) pour évaluation et autorisation.
Le Promoteur va ensuite proposer l'essai à des médecins investigateurs qui participent à l'essai et en assurent le bon déroulement dans leur établissement de soin.
L'investigateur d'un essai thérapeutique est un médecin. Il propose l'essai et informe le patient, dirige et surveille la réalisation de l'essai. Il est responsable de son déroulement et du respect des bonnes pratiques cliniques, du recueil des informations rendues anonymes et de l'archivage des données dans son centre. En cas de survenue d'un événement indésirable grave, il s'engage à informer immédiatement le promoteur qui en fera la déclaration auprès de l'ANSM. Un centre Investigateur peut être audité par le Promoteur ou les Autorités de Santé.
Le patient qui choisira de participer ou non à l'essai clinique.
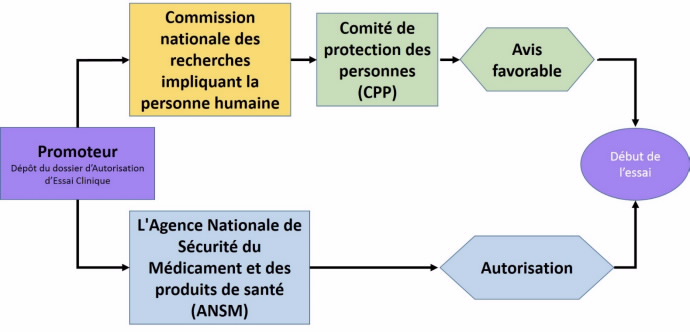
Tout promoteur d'une recherche biomédicale interventionnelle soumet le protocole de celle-ci d'une part à la commission nationale des recherches impliquant la personne humaine qui la transmets à un Comité de Protection des Personnes (CPP) désigné par tirage au sort, d'autre part à l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) qui doit délivrer une autorisation. Ce sont les deux piliers du dispositif réglementaire de contrôle de la recherche en France.
Les Comités de Protection des Personnes sont l'une des deux instances chargées d'apprécier le bien-fondé d'une recherche médicale et le respect de tous les droits des personnes appelées à y participer.
L'un des grands principes éthiques – si ce n'est le principal – de la réglementation sur la recherche médicale est que « l'intérêt des personnes qui se prêtent à une recherche prime toujours sur les seuls intérêts de la science et de la société ».
C'est pour garantir ce principe, que le législateur a créé, dès 1988, des comités de protection des personnes (CPP).
Ces comités jouent un rôle essentiel : sans un avis positif de la part de l'un d'entre eux, aucune nouvelle recherche impliquant des patients ne peut être mise en œuvre.
Depuis le décret du 16 Novembre 2016, les membres des CPP seront soumis à un programme de formation organisé par la Commission Nationale des Recherches Impliquant les personnes humaines. Les CPP seront réunis par la Commission au moins une fois par an.
L'Agence nationale de sécurité du médicament et des produits de santé, anciennement Afssaps, dont elle a repris les missions, droits et obligations. Elle a été dotée de responsabilités et de missions nouvelles, de pouvoirs et de moyens renforcés.
L'ANSM assure la gestion et l'évaluation des recherches biomédicales portant sur les produits de santé et hors produits de santé.
L'Agence évalue la sécurité et la qualité des produits utilisés au cours de la recherche, avec l'objectif de s'assurer que la sécurité des personnes se prêtant à la recherche biomédicale est garantie . » Extrait du site de l'ANSM
La commission nationale des recherches impliquant la personne humaine dépend du ministère de la santé et assure notamment la coordination et l’harmonisation du fonctionnement des CPP. Elle est composée de 22 membres nommés par arrêté du ministre chargé de la santé. Le secrétariat de la commission, assuré par la direction générale de la santé, s’occupe des échanges entre les promoteurs et les CPP. Il reçoit les dossiers des promoteurs et procède par tirage au sort à la désignation d’un CPP qui évaluera le dossier de demande d’essai clinique.
Pour en savoir plus : Décret n° 2016-1537 du 16 novembre 2016 relatif aux recherches impliquant la personne humaine
La CNIL a pour mission essentielle de protéger la vie privée et les libertés individuelles ou publiques face aux dangers que l'informatique peut faire peser sur les libertés. Elle est chargée de veiller au respect de la loi « Informatique et libertés ».
« Tout projet de traitement automatisé de données nominatives ayant pour fin la recherche dans le domaine de la santé doit faire l'objet d'une demande d'autorisation auprès de la CNIL. ».
L'objectif est de protéger les personnes contre les risques liés à la conservation et à l'utilisation des fichiers informatiques.
Source La Ligue contre le cancer
Les essais de phase I, II, III et IV (appelés phases)
Les essais de phase I correspondent le plus souvent à la première administration d'un médicament à l'homme. Les essais de phase I/II sont une variante des essais de phase I, ils permettent une évaluation préliminaire de l'efficacité à la dose sélectionnée ou bien de tester des combinaisons de médicaments.
Ils incluent en général un petit nombre de patients (10 à 40). Ces essais visent principalement à étudier la tolérance au médicament et à définir la dose et la fréquence d'administration qui seront recommandées pour les études suivantes. Un essai de phase I dure habituellement entre un et deux ans. Seuls certains services de cancérologie sont habilités à les mettre en place.
Ainsi la dose maximale tolérée, le profil de toxicité et l'activité pharmacologique du médicament seul ou parfois en combinaison avec un autre médicament, sont déterminés à l'issue de l'essai. Généralement, l'essai se déroule en deux temps : tout d'abord, une phase d'escalade de dose avec la participation d'un nombre limité de patients par palier de dose (classiquement de 3 à 6 patients), ensuite une phase d'extension au cours de laquelle plusieurs dizaines, voire une centaine de patients(partie phase II de l'essai) sont inclus pour confirmer une activité anti tumorale et la tolérance préliminaires.
L'intérêt de ces essais de phase I/II est de faciliter un accès rapide à des molécules innovantes, notamment des médicaments dits « biologiques ».
Les essais de phase II, ont pour objectif de confirmer l’activité clinique préliminaire et/ou pharmacologique du médicament à la dose recommandée à l’issue de la phase I. Un nombre limité de patients est inclus dans ces essais (40 à 80 en moyenne). Certains essais de phase II comparent deux traitements. La durée d’une phase II est généralement de deux à trois ans, dépendant de la pathologie sélectionnée et du nombre de patients.
Les essais comparatifs sont destinés à comparer le nouveau médicament à un traitement standard afin de déterminer son efficacité. Les essais de phase III incluent plusieurs centaines, voire plusieurs milliers de patients, et durent d'ordinaire au moins quatre à cinq ans, selon la pathologie et l'effet attendu.
En fonction des résultats des essais de phase III, le promoteur pourra faire une demande d'autorisation de mise sur le marché (AMM) qui permettra plus tard la commercialisation du nouveau produit.
Après leur commercialisation, les médicaments continuent à faire l'objet d'un suivi strict à long terme, dit post-AMM, afin d'identifier tout effet secondaire grave et/ou inattendu dû à son administration. On parle de pharmacovigilance. Les essais de phase IV peuvent aussi être destinés à évaluer ce nouveau médicament approuvé dans des conditions d'administration différentes, par exemple la fréquence d'administration, le nombre de cures, la durée de la perfusion…
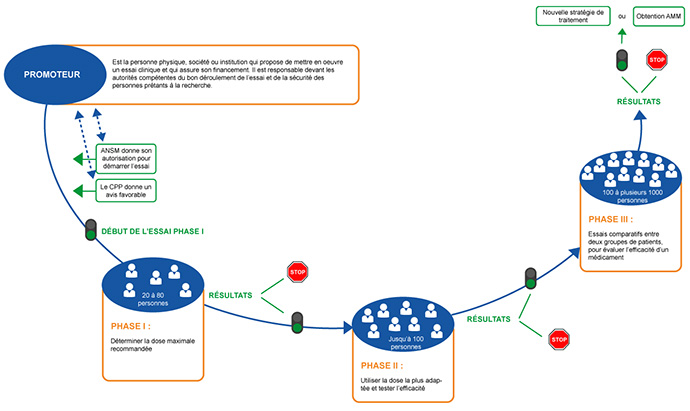
→ Attention lorsqu'on vous propose de participer à un essai clinique, celui-ci correspond à une seule phase précise (I, II ou III) du développement d'un nouveau médicament. Cela signifie que vous ne suivrez pas toutes les phases les unes à la suite des autres.
| Nombre de patients requis | Durée | But | |
|---|---|---|---|
| Phase I | 20-100 | Plusieurs mois | Sécurité / tolérance |
| Phase II | jusqu'à 100 | Plusieurs mois à 2 ans | Tolérance à court terme et surtout efficacité |
| Phase III | 100 à plusieurs 1000 | 1 à 4 ans | Evaluation et comparaisons Bénéfice / risque |
| Phase IV | Plusieurs 1000 | 1 à 4 ans | Tolérance et recherche effets indésirables |
Source La Ligue contre le cancer
Vous avez entendu parler de « recherche clinique » ou « de recherche biomédicale » ou encore « d'essais cliniques ou thérapeutiques ». Que signifient exactement ces termes ? Êtes-vous bien informés ? Cette partie à pour objectif de répondre aux questions que vous pouvez vous poser :
La recherche clinique est nécessaire pour mettre à disposition des patients de nouveaux traitements plus innovants et plus efficaces tout en faisant progresser les connaissances scientifiques sur les pathologies.
Les progrès de la médecine sont liés à l'introduction de nouveaux traitements : il peut s'agir de nouveaux médicaments, de techniques innovantes (nouveau type d'intervention chirurgicale, nouvel examen biologique, etc.) ou encore d'une nouvelle stratégie thérapeutique (combinaison de médicaments déjà connus, etc.) ou une nouvelle façon d'administrer le traitement qui sera plus confortable et/ou mieux toléré...
Avant d'être proposées à des patients en dehors des essais cliniques, ces méthodes doivent avoir fait la preuve d'une efficacité clinique, mais aussi d'une tolérance acceptable pour le patient.
Dans un premier temps, il est nécessaire de procéder pendant plusieurs mois voire des années à une longue phase expérimentale : en laboratoire d'abord, pour étudier les propriétés du médicament, de nouveaux tests biologiques pouvant inclure des tests chez l'animal, etc. On parle alors de recherche préclinique.
Dans un second temps, ces méthodes ou traitements vont être évalués chez l'Homme dans des conditions rigoureuses de sécurité, de qualité, et d'éthique : c'est la recherche clinique
Les essais cliniques ont pour objectif d'évaluer de nouveaux types de traitement ou de nouvelles stratégies thérapeutiques afin de pouvoir juger de leur efficacité et de leur utilité par rapport aux traitements actuellement disponibles. On parle de bénéfice/risque d'un traitement. Il peut aussi s'agir de techniques chirurgicales ou radiothérapeutiques différentes.
A l'origine de chaque essai clinique, il y a une question, une hypothèse scientifique de recherche.
Les résultats des essais cliniques permettent de déterminer si une nouvelle molécule ou une nouvelle stratégie thérapeutique apporte un progrès par rapport aux connaissances établies. Tous les médicaments actuellement sur le marché ont été préalablement testés et validés dans le cadre d'essais cliniques.
Cette phase d'expérimentation clinique peut être très longue, en moyenne de 2 à 5 ans. Plusieurs types d'essais (phase I, II, III) se succéderont ainsi jusqu'à ce que l'on puisse estimer que le traitement à l'essai peut être mis à disposition des patients, et, s'il s'agit de médicament, obtenir l'Autorisation de Mise sur le Marché (AMM).
Source La Ligue contre le cancer
L'Institut National du Cancer a pour mission de coordonner et d'animer la recherche clinique et translationnelle en cancérologie académique. En France, les acteurs de la recherche clinique académique en cancérologie sont notamment organisés en de multiples sociétés savantes et groupes coopérateurs. Pour permettre une meilleure efficacité dans les actions et réduire le nombre d'interlocuteurs représentatifs, l'INCA a décidé de soutenir leur regroupement et leur structuration à travers un appel à candidatures.
Actualités emploi :
Le GORTEC offre régulièrement des stages d'une durée de 6 mois, tout au long de l'année.
N'hésitez pas à envoyer vos CV et lettre de motivation en cliquant sur ce lien.
ou en envoyant un mail directement à recrutement[@]gortec.fr
Offres d'emploi :
Déposer une candidature en ligne pour les offres suivantes (CV + LM) en cliquant ici.
Actualités ORL (Bulletin Cancer)
Liste des publications :
Page 2 sur 4